Gallbladder Cancer
Carcinoid, a term first applied to hormonally active tumors by Oberndorfer in 1907,follows a more benign clinical course than most other malignancies. Carcinoid of the small intestine, a well-differentiated neuroendocrine tumor, is the most common distal small bowel malignancy, with an occurrence rate of 1 case per 300 autopsies. Carcinoid tumors of the appendix account for 0.2-0.7% of all appendicectomies, and they are the most common tumor of the appendix, accounting for 80% of appendiceal growths.2 Even though the frequency of the primary tumor is high, the incidence of metastasis is quite low (1 metastasis per 300,000 incidences). Common sites of metastatic spread include the regional mesenteric and para-aortic lymph nodes and the liver. With distant spread, especially to the liver, carcinoid syndrome can develop.
The association of flushing, diarrhea, bronchoconstriction, and cardiac disease with carcinoid tumors was first reported by Thorson and colleagues in 1954.The findings that 5-hydroxytryptamine (serotonin) was present in carcinoid tumors and that patients with carcinoid syndrome excrete increased quantities of the serotonin metabolite, 5-hydroxyindoleacetic acid (5-HIAA) led to the hypothesis that the humoral manifestations of carcinoid syndrome could be attributed to the overproduction of serotonin by these tumors. However, serotonin is not the only mediator of the clinical syndrome. Other substances, such as the tachykinins, play a significant role in the different clinical characteristics of affected patients.
Pathophysiology
Enterochromaffin cells stain yellow-brown after chromate fixation and are diffusely distributed in the tissues derived from the primitive gut. Intestinal enterochromaffin cells are the Kulchitsky cells in the crypts of Lieberkühn of the small intestine. Carcinoid tumors arise from the enterochromaffin cells. Tumor cells and Kulchitsky cells both reduce silver salts (argentaffin reaction); thus, the term argentaffinoma is used to describe carcinoid tumors.
Endocrine cells in the pituitary gland, thyroid gland, lungs, pancreas, and gastrointestinal tract secrete polypeptides and share common cytochemical and ultrastructural characteristics. Pearse developed the concept of the amine precursor uptake and decarboxylation system because these cells take up and decarboxylate amino acid precursors of biogenic amines such as serotonin and catecholamines.Included in this system are the enterochromaffin cells responsible for carcinoid tumors.
This system of cells has a common embryonic origin from the neuroectoderm. Related cells are present in the adrenal medulla, sympathetic ganglia, paraganglia, and chemoreceptor system. Because of the apparent common embryologic ancestry of these cells, a unique concept of dysplasia of the neuronal ectoderm has been proposed to explain the occurrence of multiple endocrine neoplasia and the multipotentiality of neoplastic cells derived from this system to produce a variety of peptide hormones.
Consistent with the aforementioned concept, histologic similarities among carcinoid tumors, islet cell tumors, and medullary carcinoma of the thyroid have been recognized. In addition, carcinoid tumors may coexist with other endocrine tumors. Tumors that histologically appear to be carcinoids may also produce gastrin, calcitonin, insulin, vasoactive intestinal peptide, neurotensin, catecholamines, and corticotropin (adrenocorticotropin hormone).
Common embryonic ancestry may also explain the occurrence of more than one primary carcinoid tumor in a single patient. In most instances, carcinoid tumors do not occur in association with other endocrine neoplasms, and they usually do not secrete hormones normally produced by cells other than enterochromaffin cells.
Clinical, biochemical, morphological, and cytochemical heterogeneity of carcinoid tumors are related to the site of origin. One classification is based on whether the tumor arose from the embryonic foregut (ie, bronchus, stomach, pancreas, duodenum), midgut (ie, ligament of Treitz to mid transverse colon), or hindgut (ie, mid transverse colon, descending colon, rectum). Classification can also include whether oversecretion of amine or peptide hormones is present (secretors vs nonsecretors).
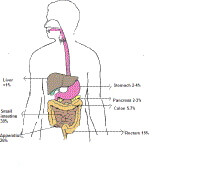
Distribution of carcinoid tumors. Characteristically, midgut-derived carcinoid tumors secrete serotonin, and patients with these tumors have elevated urinary excretion of 5-HIAA. Patients with foregut carcinoid tumors frequently have low activity of L -amino acid decarboxylase, which converts 5-hydroxytryptophan (5-HTP) to serotonin. Thus, these tumors primarily secrete 5-HTP. Midgut tumors may secrete 5-HTP in addition to serotonin. After 5-HTP is secreted, it is converted to serotonin and its metabolites by other tissues in the body. Therefore, although foregut carcinoid tumors do not usually directly secrete large quantities of serotonin, elevated urinary 5-HIAA levels are found in patients with these tumors. In contrast, hindgut carcinoid tumors do not usually secrete large amounts of either 5-HTP or serotonin, and patients with these tumors do not have elevated urinary excretion of 5-HIAA.
Endocrine aspects of carcinoid neoplasm
The term carcinoid syndrome is used to describe the hormonal manifestations of carcinoid tumors. These are flushing, diarrhea, bronchoconstriction, and cardiac disease. Most patients with carcinoid tumors do not develop carcinoid syndrome. The frequency of hormonal manifestations is greatest for midgut primary tumors and varies with the site of tumor origin. Of patients with small intestinal and proximal colonic carcinoids, 40-50% experience the syndrome. Carcinoid syndrome occurs less frequently in patients with bronchial carcinoids, is rarely observed in association with appendiceal carcinoids, and does not occur in patients with rectal carcinoids, even when the rectal carcinoid is in an advanced stage and has metastasized.
The development of carcinoid syndrome is also a function of total tumor mass and the extent of metastasis. Development is unusual in patients with a small tumor burden. Patients with the syndrome almost invariably have hepatic metastases. The association with hepatic metastases may be related to the efficient inactivation by the liver of hormones released from an abdominal tumor into the portal circulation.
In contrast, venous drainage from a metastatic tumor in the liver goes directly into the systemic circulation and bypasses hepatic inactivation. Consistent with this concept is the fact that the tumors most likely to be associated with carcinoid syndrome in the absence of hepatic metastasis are ovarian teratomas and bronchial carcinoids, which release mediators directly into the systemic circulation rather than the portal circulation.
Nonhormonal aspects
Recognition of nonhormonal symptoms early in the course of disease enhances the likelihood of diagnosis before distant metastasis or endocrine manifestations have occurred. Bronchial carcinoid tumors may be associated with respiratory complaints (eg, cough, dyspnea, hemoptysis), which leads to roentgenologic evaluation and bronchoscopy. Rectal carcinoids are usually asymptomatic in the absence of advanced disease.
Patients with midgut carcinoids frequently have symptoms for long periods (ie, 2-5 or more y) before a specific diagnosis is made. In this group of patients, early diagnosis can potentially lead to a cure by surgical resection of the primary tumor. The most common symptoms and signs of an intestinal carcinoid are abdominal pain, intermittent obstruction, and a palpable abdominal mass, each of which occurs in nearly 50% of patients.
Obstruction usually occurs after invasion of the mesentery, and the resulting desmoplastic reaction with scarring and matting of small bowel loops, in turn, can produce a mass and intermittently obstruct the intestine. The clinical picture of recurrent intermittent intestinal obstruction should raise the suggestion of carcinoid tumor. Because this process is extraluminal, results of the roentgenological examination may be normal approximately half the time.
Frequency United States
The frequency of carcinoid tumor is 1 case per 300 individuals. For carcinoid syndrome, it is 1 case per 300,000 individuals.
About 11,000 to 12,000 neuroendocrine tumors and neuroendocrine cancers are diagnosed each year in the United States. About 2 out of 3 of these occur in the digestive system.5
Mortality/Morbidity
Survival is dependent on the primary site and the size of the primary tumor. Patients with metastatic disease arising from midgut (ie, distal small intestine/cecal) primary tumors generally live longer than those with the foregut (ie, bronchial, gastric, pancreatic) and hindgut (ie, rectal) as primary sites. The 5-year survival rate from the time of diagnosis of metastatic disease is 67%. No therapy to date has been shown in any randomized clinical trial to prolong survival for patients with metastatic carcinoid tumors, and therapy remains palliative.
Free Consultation
Conditions